To truly harness the power of quantitative proteomics in order to understand complex biological phenomena, researchers require accurate and high-throughput methods capable of comprehensively quantifying the large numbers of proteins present in biological samples. Isobaric chemical tagging can be used to quickly determine relative changes in multiple complex protein samples in a single analysis. Used in combination with the latest ultra-high resolution mass spectrometry (MS) techniques, these approaches enable proteomic analysis of very large numbers of samples simultaneously, granting access to comprehensive proteome-wide quantitation faster and more accurately than ever before.
This article details how these techniques have been used in a study at Massachusetts General Hospital and Harvard Medical School, to characterize the interactome for Xist, a noncoding RNA responsible for silencing the inactive female X chromosome (Xi). It’s hoped that by identifying which interacting protein partners repress Xi transcription, such knowledge will help other researchers more fully understand gene silencing on a global scale.
Understanding gene silencing
Most cells in the human body contain essentially the same genetic material. So why do we have so many different cell types and how can they perform such a broad range of functions? What separates a red blood cell, for instance, from a nerve cell —and the majority of metabolic pathways involved in each—is the extent to which certain genes are either expressed or silenced.
Put simply, differential gene expression is what makes us tick, and being able to control when and how genes are expressed could form the basis of therapies for many diseases, including neurodegenerative disorders or cancer. Yet identifying which genes are inactivated or silenced is not a simple task, and cannot be done simply by sequencing the genetic code. This is because a number of the factors affecting gene expression are epigenetic in nature, meaning they are not intrinsically linked to the genome itself. Whilst the mechanisms for gene silencing are not fully known, it is understood that the regulation of gene expression often depends on interactions between RNA molecules and proteins.
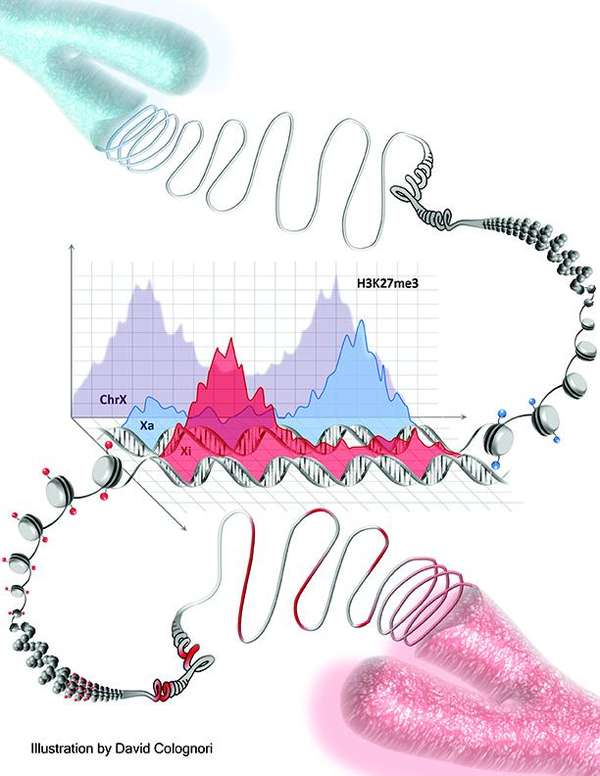
Due to its large size—around five percent of a cell’s genetic material —the female Xi chromosome is an excellent model to study gene silencing. Female somatic cells possess two X chromosomes, while males have only one. In order to prevent the lethal overproduction of RNA and proteins that would occur by having a second, transcriptionally-active X chromosome, one of the X chromosomes is permanently inactivated during early development in the womb. A gene known as Xist, present on both X chromosomes but expressed only on Xi, is responsible for producing several hundred copies of a noncoding RNA molecule that bind to the Xi chromosome and associate with proteins in order to inactivate it. Therefore, to better understand the mechanism of Xi inactivation, it is essential to understand with which protein Xist RNA interacts. The quest to identify the “proteome” for Xist RNA extended 25 years and only recently came to an end. The proteome identified in 2015 was made possible by two advances: iDRiP, a novel method of purifying RNA-protein interactions; and the multiplex proteomics TMT SPS MS3.
Mapping the Xist protein interactome
One of the challenges associated with conventional MS proteomics experiments is throughput. With some techniques, analysis of multiple affinity-enriched protein samples requires individual MS experiments. The approach is time-consuming and important proteins may not be quantified in each of the individual experiments, which complicates the interpretation of the results. Confidently and comprehensively mapping the large numbers of proteins in this investigation using replicate experiments under multiple experimental conditions, therefore, requires the latest high-throughput techniques to facilitate rapid and comprehensive analysis.
Multiplex proteomics approaches using isobaric chemical tagging techniques such as tandem mass tag (TMT) labeling allow for the identification and quantification of relative changes in complex protein samples across multiple conditions simultaneously. When mapping the Xist interactome, replicate samples were labeled with TMT reagents and compared to each other in a single experiment on an Orbitrap Fusion mass spectrometer, as reported in the October 2016 issue of the journal Science. The TMT10plex mass tag labeling approach allows up to 10 separate samples to be investigated, each with its own unique reporter ions. Following fragmentation of the precursor ions, the relative levels of each protein are quantified by comparing the amount of detected reporter ions generated from each tag. The high-throughput nature of the approach means that more than a hundred affinity-enriched samples can be compared in the space of a week.
Key to the success of the analysis was the MS approach employed. Isobaric labeling multiplexing approaches based on conventional tandem MS can be problematic, as the presence of co-eluting and co-fragmenting isobaric species – even at low levels – can mask the precursor ion signals of interest. This causes suppression of protein ratios and loss of quantitative accuracy and precision. The use of an additional MS3 analysis stage in combination with synchronous precursor selection (SPS) overcame the problem of isobaric ion contamination to ensure accuracy of peptide quantitation whilst maintaining sensitivity in these low concentration pull down samples.
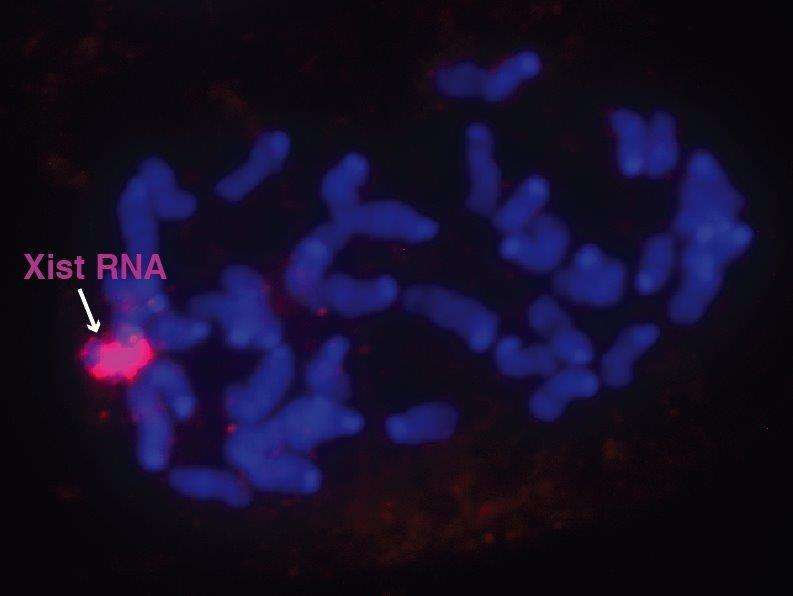
Regulating gene inactivation
Using the TMT SPS MS3 method, over 200 proteins found to interact with Xist were identified. These included protein complexes known to play a role in X chromosome inactivation, such as polycomb group protein PCR2, as well as a large number of proteins from multiple categories that were previously not known to interact with Xist. Many of these epigenetic complexes comprised multiple protein sub-units, boosting confidence in their role as Xi interactors. Of particular interest was the identification of a number of cohesion proteins sub-units —a class of architectural proteins responsible for changing chromosome shape.
By treating cell lines with commercially available protein inhibitors and gene-specific biologic compounds, partial reactivation of the Xi chromosome was observed. However, in order to get good perturbation of the inactive accent, at least two or three proteins had to be targeted, suggesting that Xi silencing is a particularly robust process involving multiple parallel mechanisms.
Results of the cohesion reactivation studies, for instance, revealed surprising insight into the role these proteins play. These experiments found that Xist actually works to repel the action of these architectural proteins on the Xi chromosome, rather than recruiting their presence to make the chromosome transcriptionally inactive.
Conclusion
In order to perform large-scale proteomics investigations involving multiple complex samples, high-throughput methods capable of accurately and rapidly generating protein characterization data are essential. In this study, the latest tandem mass tag labelling and synchronous precursor selection mass spectrometry techniques were used to fully characterize the Xist protein interactome. Such technology is bringing about new and surprising insight into important biological pathways that may ultimately be used to find innovative therapeutic treatments.
Additional Materials