The Role of Cryo-TEM for Structural Biology - Part I
 Editor's Note: This is the first of a three-part series by Thomas Wohlfarth discussing the role of Cryo-TEM for structural biology.
Editor's Note: This is the first of a three-part series by Thomas Wohlfarth discussing the role of Cryo-TEM for structural biology.
All living organisms are vastly complex assemblies consisting of organs, tissues, cells, organelles and molecular protein assemblies. It all starts with the genes. Those hold the information for the proteins they are coding for, and after translation of the genetic information, those proteins are key for many biochemical and physiological processes that determine how cells and tissues develop and differentiate. However, the majority of those proteins do not execute their function as single entities, but act as protein complexes. Understanding their structure and deducing their function is absolutely key to unraveling the fundamentals of life and, subsequently, also for developing smarter drugs to fight today’s and tomorrow’s diseases.
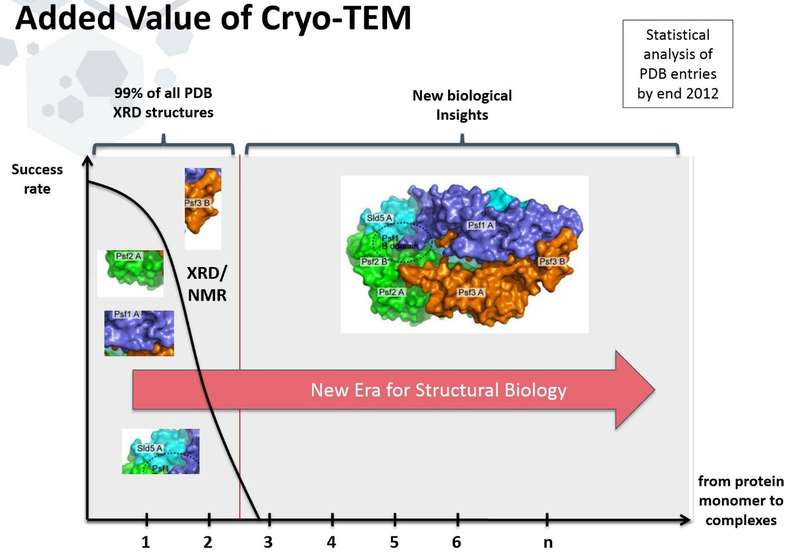
Since the human genome was decoded a decade ago, we have known the primary sequence of every protein in the human body. Since then, the genomes of many other organisms have also been decoded. However, these sequences alone do not tell us the shape those proteins will assume in order to perform their functions. Using tools like x-ray crystallography (XRD) and nuclear magnetic resonance (NMR), we have been able to determine the structures of many proteins down to the atomic scale, but these tools each have limitations that prevent them from analyzing most proteins let alone their complexes. NMR analysis is mainly focusing on parts of a single protein or protein domains (typically less than 35 kDa, although larger structures have been solved). XRD, on the other hand, can only be applied to the limited number of proteins that can be coaxed to form regular crystals — a limitation that excludes many biologically-important proteins, and precludes any observations of dynamic behavior.
Cryo-electron microscopy (cryo-EM) offers the opportunity to look at a wide range of biological structures while being in a native hydrated state. It visualizes proteins and proteins complexes in the “closest to reality” state. In addition, structural analysis by using cryo- EM does not require the molecules to be crystallized and is not restricted by the size of the molecules. Cryo-EM structural analysis now delivers resolutions in the sub 3 Ångstroms range -- sufficient to reveal tertiary and quaternary protein structure and, in some cases, to identify individual amino acid side chains. Using powerful computational methods, researchers can often fit atomic scale results from smaller component structures derived by XRD and NMR, into larger scale structures determined by cryo-EM.
This three-part article discusses how biologists are adopting a powerful new approach to structural analysis that uses sophisticated computational tools to integrate molecular-scale information from EM with atomic-scale results from XRD and NMR. This new approach, known as Integrated Structural Biology, enables scientists to visualize and understand the relationship between structure and function in the tiny molecular machines that constitute all living systems. Integrated Structural Biology overcomes the limitations of the individual techniques to allow researchers to gain ground-breaking new insights into the way living systems function, or malfunction, and may suggest promising pathways for the development of effective new medicines and other molecular therapies.
The next article of this series will take a closer look at cryo-EM: how it works and its benefits and limitations.