The Role of Cryo-TEM for Structural Biolog - Part II
Editor's Note: This is the second of a three-part series by Thomas Wohlfarth discussing the role of Cryo-TEM for structural biology.
In part I of my article last week, I presented the concept of Integrated Structural Biology that combines XRD and NMR data that is already available with cryo-TEM to gain new biological insights. Now in part II, we will take a closer look at cryo-TEM: how it works as well as its benefits and limitations.
A transmission electron microscope (TEM) operates according to many of the same principles used in a light microscope. An electron source generates a beam of high energy electrons that interacts with a part of the sample. The majority of electrons interact with sample atoms and upon transmission through the sample are focused by electromagnetic lenses onto an imaging device such as a charged coupled device (CCD) or direct electron detector (DED). The path travelled by the beam, from source to camera, must be kept under vacuum to prevent the scattering of electrons by gas molecules and consequently unwanted ionization effects in the column.
Biological samples present several challenges to the electron microscopist. Biological materials are generally composed of relatively light elements and do not generate as much imaging contrast as materials composed of heavier elements. This might be addressed by either longer exposure times, lower accelerating voltages or applying stronger defocus values. In general, biological materials are typically quite susceptible to damage by the high energy electrons of the beam. Finally, the samples must tolerate the vacuum environment of the sample chamber and should not become contaminated due to the vacuum regime of the microscope.
Cryogenic sample preparation techniques address the vacuum requirements by solidifying the liquid water found in most biological materials. The samples are typically prepared in an aqueous suspension that is plunge frozen in a bath of liquid ethane and or propane. The freezing process is so rapid that the water does not crystallize as cubic or hexagonal structure, but instead forms an amorphous solid that preserves the fine structure of the sample in a nearly natural state. As of today, there are various commercial systems available (such as the FEI Vitrobot) to automate the cryo preparation process and maintain careful control of all critical parameters.
To address the inherently low contrast of biological specimens without introducing stains that can distort structures and cause other artifacts, microscopists use computational techniques to combine multiple images of identical particles. In the composite image, the signal-to-noise ratio increases as the signal sums while the background averages out to a constant, low level. Great care must be taken throughout the imaging process to avoid excessive exposure of the sample to the beam.
Single particle analysis (SPA) and electron tomography (ET) are two different techniques that combine multiple images to build contrast and provide three dimensional structural analysis.
Workflow
TEMs have evolved dramatically from the complex, difficult to use “science projects” of their early years, to sleek, highly automated, easy-to-use instruments. Automation not only makes the system easier to operate, it provides a much higher degree of repeatability and enables lengthy acquisitions of very large data sets without operator intervention. System stability is also a requirement for long acquisitions. System designers have made every effort to develop a TEM workflow that blends seamlessly with the specific needs of the systems users. For biological applications, this includes optimization and stabilization of the sample in an appropriately buffered suspension, cryogenic sample preparation, acquisition of the image/data set, data/image analysis, model reconstruction and visualization. The system is designed to allow biologists to seek answers to biological questions without becoming expert microscopists.
Cryo-TEM Examples
Non-enveloped Virus Priming Mechanism for Cell Entry
To achieve cell entry, many nonenveloped viruses must transform from a dormant to a primed state. In contrast to the membrane fusion mechanism of enveloped viruses (e.g., influenza virus), this membrane penetration mechanism is poorly understood. Here, using single-particle cryo-electron microscopy, the authors report a 3.3 Å structure of the primed, infectious subvirion particle of aquareovirus. The density map reveals side-chain densities of all types of amino acids (except glycine), enabling construction of a full-atom model of the viral particle. The structure and biochemical results show that priming involves autocleavage of the membrane penetration protein and suggest that Lys84 and Glu76 may facilitate this autocleavage in a nucleophilic attack. They observe a myristoyl group, covalently linked to the N terminus of the penetration protein and embedded in a hydrophobic pocket. These results suggest a well-orchestrated process of nonenveloped virus entry involving autocleavage of the penetration protein prior to exposure of its membrane-insertion finger.
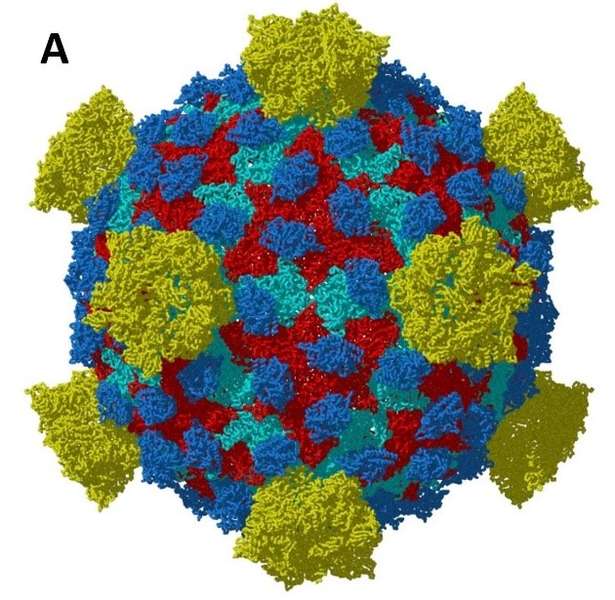
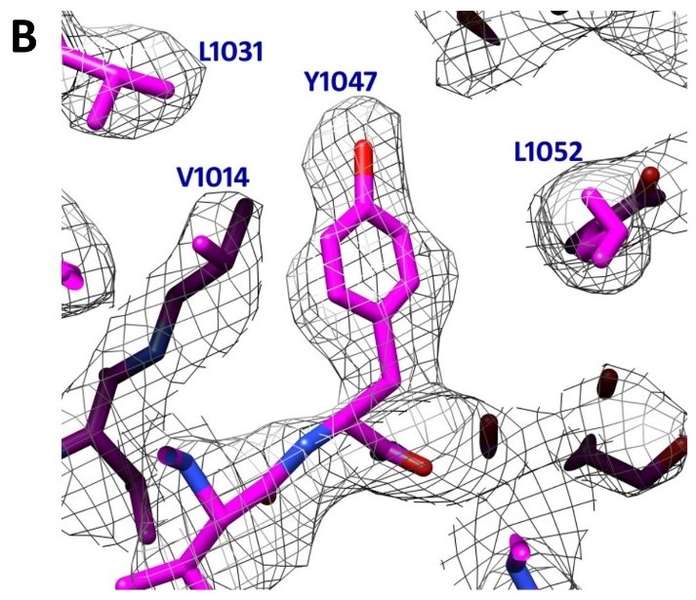
Fig. 1. A 3.3 Å structure of a non-enveloped icosahedral virus using Cryo-TEM. B Image of a selected region were individual side chains are clearly visible in the Cryo-TEM reconstruction validating the high resolution of the reconstruction. Images kindly provided by Z. Hong Zhou Electron Imaging Center for Nanomachines (EICN) CNSI and Department of Microbiology, Immun & Mol. Genetics, UCLA. Adapted from [2]. EMDB Accession code: EMD-5160; Fitted PDB ID: 3IYL
Trimeric HIV-1 Envelope Glycoprotein Activation
HIV-1 infection begins with the binding of trimeric viral envelope glycoproteins (Env) to CD4 and a co-receptor on target Tcells. Understanding how these ligands influence the structure of Env is of fundamental interest for HIV vaccine development. Using cryo-electron microscopy, the authors describe the contrasting structural outcomes of trimeric Env binding to soluble CD4, to the broadly neutralizing, CD4-binding site antibodies VRC01, VRC03 and b12, or to the monoclonal antibody 17b, a co-receptor mimic. Binding of trimeric HIV-1 BaL Env to either soluble CD4 or 17b alone, is sufficient to trigger formation of the open quaternary conformation of Env. In contrast, VRC01 locks Env in the closed state, while b12 binding requires a partial opening in the quaternary structure of trimeric Env. The results show that, despite general similarities in regions of the HIV-1 gp120 polypeptide that contact CD4, VRC01, VRC03 and b12, there are important differences in quaternary structures of the complexes these ligands form on native trimeric Env, and potentially explain differences in the neutralizing breadth and potency of antibodies with similar specificities. From cryo-electron microscopic analysis at, 9 Å resolution of a cleaved, soluble version of trimeric Env, they show that a structural signature of the open Env conformation is a three-helix motif composed of α-helical segments derived from highly conserved, non-glycosylated N-terminal regions of the gp41 trimer. The three N-terminal gp41 helices in this novel, activated Env conformation are held apart by their interactions with the rest of Env, and are less compactly packed than in the post-fusion, six-helix bundle state. These findings suggest a new structural template for designing immunogens that can elicit antibodies targeting HIV at a vulnerable, pre-entry stage.
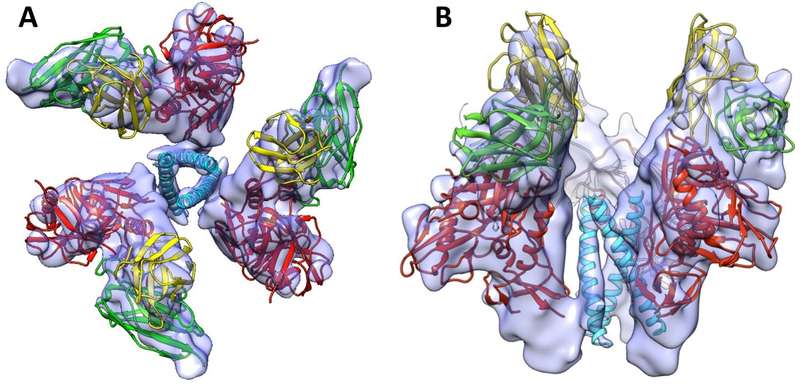
Fig. 2 9 A° three-dimensional reconstruction of soluble gp140 HIV envelope glycoprotein trimers bound to three copies of the Fab fragment from 17b, a neutralizing antibody whose binding mimics that of the co-receptor. The structure revealed the presence of a previously unknown ‘‘activated’’ intermediate state, where three buried helices become exposed and potentially accessible to binding by entry inhibitors. A Top view of the trimer and B Side view of the trimer. Images kindly provided by Sriram Subramaniam, Lab of Cell Biology, National Cancer Institute, National Institutes of Health. Adapted from [3]. EMDB Accession code: EMD-546
HIV-1 Capsid Structure
Retroviral capsid proteins are conserved structurally but assemble into different morphologies. The mature human immunodeficiency virus-1 (HIV-1) capsid is best described by a fullerene cone model, in which hexamers of the capsid protein are linked to form a hexagonal surface lattice that is closed by incorporating 12 capsid-protein pentamers. HIV-1 capsid protein contains an amino-terminal domain (NTD) comprising seven a-helices and a b-hairpin, a carboxy-terminal domain (CTD) comprising four a-helices, and a flexible linker with a 310-helix connecting the two structural domains. Structures of the capsid-protein assembly units have been determined by X-ray crystallography; however, structural information regarding the assembled capsid and the contacts between the assembly units is incomplete. Here we report the cryo-electron microscopy structure of a tubular HIV-1 capsid-protein assembly at 8 Å resolution and the three-dimensional structure of a native HIV-1 core by cryo-electron tomography. The structure of the tubular assembly shows, at the three-fold interface, a three-helix bundle with critical hydrophobic interactions. Mutagenesis studies confirm that hydrophobic residues in the center of the three-helix bundle are crucial for capsid assembly and stability, and for viral infectivity. The cryo-electron-microscopy structures enable modelling by large-scale molecular dynamics simulation, resulting in all-atom models for the hexamer-of-hexamer and pentamer-of hexamer elements as well as for the entire capsid. Incorporation of pentamers results in closer trimer contacts and induces acute surface curvature. The complete atomic HIV-1 capsid model provides a platform for further studies of capsid function and for targeted pharmacological intervention.
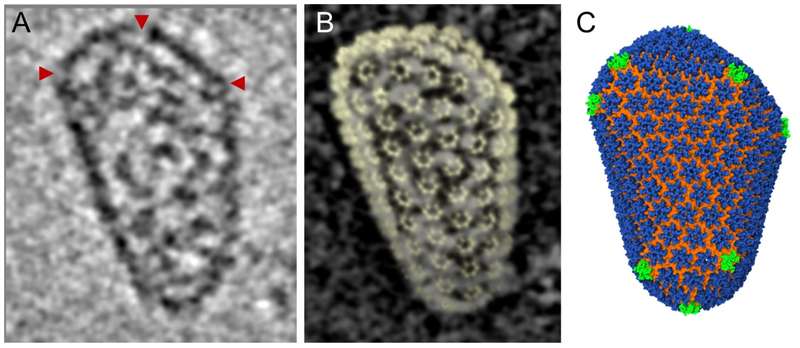 Fig. 3 Using cryo-tomographic analysis of individual, native ‘‘fullerene’’ cone HIV-1 capsid, combined with the high resolution Cryo-TEM hexamer structures an all-atom molecular dynamics HIV-1 capsid model was created. This model highlights the threefold capsid protein C-terminal domain as an attractive therapeutic target. A Slice through cryo-tomographic volume of an individual mature capsid. Red arrows highlight CA pentamers. B The tomographic density matches the shape and size of the capsid, shown by the overlay of densities from the segmented capsid and the fullerene model (yellow). C Final molecular dynamics equilibrated all-atom capsid model. Images kindly provided by Peijun Zhang Univ. of Pittsburgh. Adapted from [6] EMDB Accession codes: EMD-5582, EMD-5639; Fitted PDB IDs: 3J34, 3J4F, 3J3Y
Fig. 3 Using cryo-tomographic analysis of individual, native ‘‘fullerene’’ cone HIV-1 capsid, combined with the high resolution Cryo-TEM hexamer structures an all-atom molecular dynamics HIV-1 capsid model was created. This model highlights the threefold capsid protein C-terminal domain as an attractive therapeutic target. A Slice through cryo-tomographic volume of an individual mature capsid. Red arrows highlight CA pentamers. B The tomographic density matches the shape and size of the capsid, shown by the overlay of densities from the segmented capsid and the fullerene model (yellow). C Final molecular dynamics equilibrated all-atom capsid model. Images kindly provided by Peijun Zhang Univ. of Pittsburgh. Adapted from [6] EMDB Accession codes: EMD-5582, EMD-5639; Fitted PDB IDs: 3J34, 3J4F, 3J3Y
Cryo-EM has evolved into a useful tool for biology in the past decade. The final part of this article series will discuss the combined value of NMR, XRD and cryo-EM, known as Integrated Structural Biology.