Tumors Dedifferentiated by Chemo May Spawn New Paradigm
 A Harvard University and Waterloo University team has found that a common breast cancer chemotherapy (chemo) can create stem-like cancer cells out of more differentiated tumor cells.
A Harvard University and Waterloo University team has found that a common breast cancer chemotherapy (chemo) can create stem-like cancer cells out of more differentiated tumor cells.
The team then applied a therapy that targets stem-like cancer cells—and killed off the cells.
This paper, with a handful of others, may explain many kinds of mysterious chemo resistance. It could also usher in a new kind of cancer therapy, in which clinicians deliberately shoot for a one-two punch; deliberately create new kinds of cancer cells out of tumors—to better kill them.
“This research shows us yet another sense in which cancer is a moving target,” Charlotte Kuperwasser, Ph.D., uninvolved in the study, told Bioscience Technology. Kuperwasser is director of the Tufts University Laboratory for the Convergence of Biomedical, Physical and Engineering Sciences. “It also shows us how therapies can be rationally designed and scheduled so that they stay one step ahead of a cancer cell’s ability to shift between phenotypic states.”
Chemo’s stress may create stem-like cells
The Harvard team, led by Brigham Women’s Hospital associate bioengineer Shiladitya Sengupta, Ph.D., set out to analyze resistance to chemo. Much prior work has postulated that cancer stem cells (CSCs), often considered the most long-lasting, robust, and thus deadly cells in cancer, can persist despite chemo—then spring to life later.
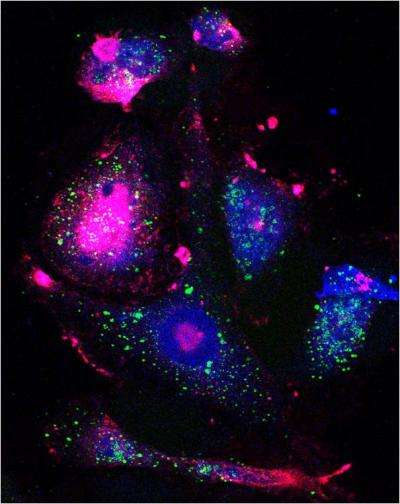
In Nature Communications, Sengupta’s team reported a different thing happened when they exposed breast cancer cells, which didn’t express CSC markers, to docetaxel. After docetaxel exposure, the differentiated cancer cells began expressing some CSC markers. These “markers of stemness" indicated the cells had transitioned into a more dedifferentiated state—which might now make them vulnerable to a different class of anti-cancer drugs.
The team had apparently created cancer stem-like cells out of more differentiated cancer cells (if the changed cells did not express all expected stem cell markers).
The team then treated the newly altered cells with new therapies. Two drugs each killed many of the transitioning cells. The two drugs were dasatinib, which targets the Src Family Kinase (SFK) (and is known to kill progenitor and stem-like cancer cells), and RK20449, a new drug in pre-clinical testing that targets an SFK protein called Hck (oft expressed in embryonic stem cells).
The team confirmed this in mice with mammary carcinoma. Treatment with dasatinib—a few days after administering two high doses of docetaxel—halted tumor growth, and increased mouse survival rates (presumably as this was the time it took the cells to dedifferentiate). Treating cells with docetaxal and dasatinib at the same time did not produce that effect. Administering dasatinib after a longer period of time also failed to work (presumably because the cells had begun differentiating normally again.)
The team used mathematical modelling, provided by Waterloo researchers, to help predict effective chemo doses.
 Lead author Aaron Goldman Ph.D., a postdoctoral fellow at BWH, told Bioscience Technology he did not think what he saw was an example of the epithelial-to-mesenchymal (EMT) transition used by some to explain some transitions to more virulent cancer states. “I don’t believe what we see is tantamount to an EMT,” he said. “Classically, the markers which designate a mesenchymal phenotype are predicated on a CD44HiCD24Lo biomarker expression profile. We found cells induced by the therapy will alter their phenotype towards a CD44HiCD24Hi trajectory. In the classical sense, this may be more closely related to a mesenchymal to epithelial transition (MET). “
Lead author Aaron Goldman Ph.D., a postdoctoral fellow at BWH, told Bioscience Technology he did not think what he saw was an example of the epithelial-to-mesenchymal (EMT) transition used by some to explain some transitions to more virulent cancer states. “I don’t believe what we see is tantamount to an EMT,” he said. “Classically, the markers which designate a mesenchymal phenotype are predicated on a CD44HiCD24Lo biomarker expression profile. We found cells induced by the therapy will alter their phenotype towards a CD44HiCD24Hi trajectory. In the classical sense, this may be more closely related to a mesenchymal to epithelial transition (MET). “
On the other hand, he said, “I would be hesitant to characterize the cells in these behavior patterns because I believe it diminishes the heterogeneity of cancers and their response to therapies. Tumors are composed of genetic and phenotypic diversity in which their distinct phenotypes are in constant flux, and dynamic equilibrium, as a consequence of inherent phenotypic plasticity. I believe that, in the context of drug resistance, classifying cells into a hierarchy is somewhat impeding. It seems more rational, based on our evidences, to visualize them on a continuum in which dynamism exists in a back-and-forth direction depending on what specific features are requisite for survival. Of note, we identified that a reversion of the phenotype is achieved over time, suggesting that once cells acquire a new state—whether that can be defined as MET or not—they achieve the old heterogeneity following cessation of treatment. Does that mean they undergo an ‘EMT’ at some point during their transient transitioning? I am not certain of that.”
Some other groups—including the group of University of Michigan oncologist Max Wicha, M.D.,—have postulated that resistant cancer cells left over, post-chemo, tend to be robust cancer stem cells that were there from the start, but were undetected, and were not killed because they were resistant to chemo. One postulated reason: stem cells are quiescent, and many chemos work on actively replicating cells.
Goldman says his group is postulating something very different.
“Dr. Wicha’s lab has done some very interesting research. I am not certain that these models--our evidence for phenotypic state-transitions, versus his model of CSCs—necessarily stack-up together.” One Wicha paper postulates that many luminal cancers first thought to be HER-2 negative—not containing stem-like cells—turn out to possess undetected stem-like cells expressing HER-2. They replicate when the cancers return post-chemo. Wicha’s lab has suggested that all patients with luminal cancer may benefit from Herceptin, which targets HER-2.
“That paper is quite fascinating in that subpopulations of HER-2 positive cells may exist in HER-2 negative breast cancers to drive growth potential. They posit that a static population of CSC existing prior to therapy can be targeted, ablated, and potentiate lower tumor growth incidence. In contrast, our study emphasizes cancer cell plasticity as greatly important to the sub-clonal heterogeneity of tumors. Therapy can exacerbate this plasticity, forcing cells into new phenotypic states. I think classifying cancer cells into hierarchical arrangements may need to be dissolved in models of adaptive resistance to chemotherapy.”
Can chemo-driven plasticity into a stem cell like state explain most resistance? “This question is quite interesting,” he told Bioscience Technology. “I believe that researchers have hit on a very interesting notion, that certain phenotypes can create a tumor initiating, or drug resistant, capacity with other malignant features. It has been attractive to classify malignant phenotypes as CSCs. CSCs are unfortunately poorly defined by specific markers and designations since so much controversy and disagreements exist. For example, even our recent study determined that inherent (drug naïve) CD44HiCD24Hi cells will induce tumors much more quickly than CD44HiCD24Lo (the classic breast CSC phenotype).”
Still, he said, he finds it fascinating that in classical CSC research, “particular phenotypes are ‘favored’ and that transitioning into this favored phenotype reveals certain malignant characteristics. Antonija Kreso and John Dick published a fascinating study in Science recently that also highlights the dynamism of subclonal heterogeneity in therapy response.”
Ultimately, Goldman told Bioscience Technology, “I believe transitioning into a ‘favored’ state will reveal much of the cause of resistance to chemotherapies. It will be critical, I believe, to find what properties allow certain cells to acquire these features. Does a certain genetic predisposition or epigenetic profile allow a cell to gain ‘favored’ properties? That will be fascinating to uncover if we seek to target adaptive resistance.”
Kuperwasser concluded to Bioscience Technology: “It will be important to see if similar strategies can be explored in other cancer models and tumor types that take into account cancer cell plasticity.”