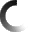The Role of Cryo-TEM for Structural Biology - Part III
Editor's Note: This is the final installment of a three-part series by Thomas Wohlfarth discussing the role of Cryo-TEM for structural biology.
In part one of this series, I presented the concept of Integrated Structural Biology that combines XRD and NMR data that is already available with cryo-TEM to gain new biological insights. Part two explored cryo-TEM in-depth: how it works, its benefits and limitations. In the final part of this series, I will discuss the value of Integrated Structural Biology with a few examples.
In 2005 Wanker and colleagues analyzed the human protein-protein interaction network using an automated yeast two hybrid system. They found a large, highly connected network comprising almost 3186 novel interactions among 1705 proteins. Of these, they identified 911 high-confidence interactions among 401 proteins. The vast majority of protein interactions involve dynamic protein complexes between 50 and 500 kDa. Most proteins work together, somewhat like the parts of a car. To continue that analogy, we could not hope to understand how a car works by examining the parts in isolation. We need to see how they work together. In the same way we need to see how proteins work together to understand biological systems. For this we need the fine resolution of NMR and x-ray crystallography to look at the parts and the larger scale view of cryo-EM to fit them together.
The protein data base (PDB) had some 90,000 submission in 2013. 99% of all crystallographic PDB structures have less than two components. In the same year, 711 EM structures were submitted. While this number is small in comparison to the number of XRD structures, it has grown nearly tenfold over the preceding decade and the growth rate is accelerating quickly. There is a very large uncharted space for large complexes, space where important biological questions remain to be answered (see diagram in Part I of this series). Integrative approaches can combine the XRD and NMR data that is already available with cryo-TEM to gain new biological insights. Here we will present a few examples of the value of Integrated Structural Biology.
Electron Microscopy Structure of Human APC/CCDH1-EMI1 Reveals Multimodal Mechanism of E3 Ligase Shutdown
The Anaphase Promoting Complex/Cyclosome (APC/C) is a ~1.5 MDa multiprotein E3 ligase enzyme that regulates cell division by promoting timely ubiquitin-mediated proteolysis of key cell cycle regulatory proteins. Inhibition of human APC/CCDH1 during interphase by Early Mitotic Inhibitor 1 (EMI1) is essential for accurate coordination of DNA synthesis and mitosis. Here, we report a hybrid structural approach involving NMR, electron microscopy, and enzymology, which reveal that EMI1’s 143-residue C-terminal domain inhibits multiple APC/CCDH1 functions. The intrinsically disordered D-box, Linker, and Tail elements, together with a structured zinc-binding domain, bind distinct regions of APC/CCDH1 to synergistically both block the substrate-binding site and inhibit ubiquitin chain elongation. The functional importance of intrinsic structural disorder is explained by enabling a small inhibitory domain to bind multiple sites to shut down multiple functions of a “molecular machine” nearly 100 times its size.
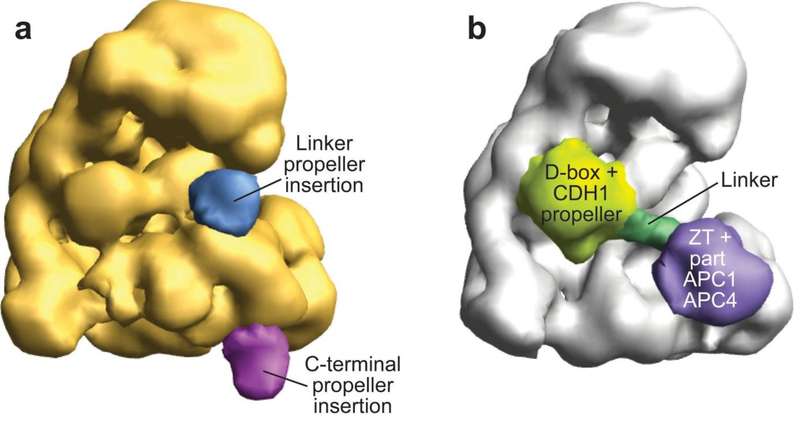
Aerolysin Pore Membrane Insertion Mechanism
Aerolysin is the founding member of a superfamily of β-pore–forming toxins whose pore structure is unknown. The authors have combined X-ray crystallography, cryo-EM, molecular dynamics and computational modeling to determine the structures of aerolysin mutants in their monomeric and heptameric forms, trapped at various stages of the pore formation process. A dynamic modeling approach based on swarm intelligence was applied, whereby the intrinsic flexibility of aerolysin extracted from new X-ray structures was used to fully exploit the cryo-EM spatial restraints. Using this integrated strategy, they obtained a radically new arrangement of the prepore conformation and a near-atomistic structure of the aerolysin pore, which is fully consistent with all of the biochemical data available so far. Upon transition from the prepore to pore, the aerolysin heptamer shows a unique concerted swirling movement, accompanied by a vertical collapse of the complex, ultimately leading to the insertion of a transmembrane b-barrel.
This three-part article has reviewed the use of a powerful new approach to structural analysis that uses sophisticated computational tools to integrate molecular-scale information from a cryo-TEM with atomic-scale results from XRD and NMR. With this new approach, known as Integrated Structural Biology, scientists are able to visualize and understand the relationship between structure and function in the tiny molecular machines that constitute all living systems. This deeper understanding gives scientists the potential to unraveling the fundamentals of life and develop new, better drugs for the future.